11. phaseshifts API
11.1. Package Contents
This chapter covers the main modules of the phaseshifts and provides some API documentation for those wishing to incorporate this package into their own projects.
11.2. Subpackages
- The main sub packages are listed below:
phaseshifts.gui- includes all the necessary files for the graphical user interface.phaseshifts.lib- contains the Fortran libphsh library and the python wrappings.phaseshifts.doc- source documentation for the phaseshifts package.phaseshifts.test- modules for testing the phaseshift package.
11.3. Submodules
11.3.1. phaseshifts.atorb
atorb.py
This module provides a high-level Python interface for generating input files and executing atomic structure calculations using the ‘atorb’ program (part of the Barbieri/Van Hove LEED phase shift package).
The core functionality allows users to:
1. Determine the ground-state electronic configuration of elements.
2. Construct formatted input files for the atorb Fortran solver.
3. Calculate radial charge densities 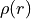 by solving the
by solving the
Dirac-Fock (relativistic) or Hartree-Fock (non-relativistic) equations for a free atom.
These charge densities serve as the starting potential for calculating scattering phase shifts in Low-Energy Electron Diffraction (LEED) and photoelectron diffraction experiments.
- See:
http://www.icts.hkbu.edu.hk/surfstructinfo/SurfStrucInfo_files/leed/
- Requires:
f2py (for libphsh fortran wrapper generation)
Note
To generate libphsh fortran wrappers (libphsh.pyd) for your platform then use ‘python setup.py’ in the lib directory of this package to install into your python distribution. Alternatively, use:
f2py -c -m libphsh libphsh.f
Windows users may have to add appropriate compiler switches, e.g.
# 32-bit
f2py -c -m libphsh --fcompiler=gfortran --compiler=mingw-32 libphsh.f
# 64-bit
f2py -c -m libphsh --fcompiler=gfortran --compiler=mingw-64 libphsh.f
- class phaseshifts.atorb.Atorb(ngrid=1000, rel=True, exchange=0.0, relic=0, mixing_SCF=0.05, tolerance=0.0005, xnum=100, ifil=0, **kwargs)[source]
Bases:
objectWrapper for the atorb atomic structure solver.
This class encapsulates the logic for interacting with the Barbieri/Van Hove atorb program. It calculates atomic orbitals and radial charge densities on a logarithmic grid.
The solver numerically integrates the radial Dirac equation (or Schrödinger equation if relativistic effects are disabled) to find the eigenvalues and eigenfunctions of the bound electrons.
Notes
The Breit interaction is neglected, which is a valid approximation for valence states dominating the scattering potential in LEED.
Original author: Eric Shirley
There are nr grid points, and distances are in Bohr radii
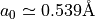
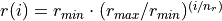 ,
,
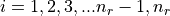
The orbitals are stored in phe(), first index goes
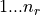 , the
second index is the orbital index (
, the
second index is the orbital index (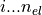 )
)Look at the atomic files after printing this out to see everything… Suffice it to say, that the charge density at radius
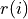 in units of electrons per cubic Bohr radius is given by:
in units of electrons per cubic Bohr radius is given by: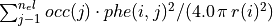
Think of the phe functions as plotting the radial wave-functions as a function of radius on a logarithmic mesh…
The Dirac equation is solved for the orbitals, whereas their density is treated by setting
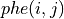 to Dirac’s
to Dirac’s
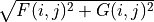 times the sign of
times the sign of 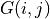 …
…So we are doing Dirac-Fock, except that we are not treating exchange exactly, in terms of working with major and minor components of the orbitals, and the phe’s give the CORRECT CHARGE DENSITY…
The above approximation ought to be very small for valence states, so you need not worry about it…
The Breit interaction has been neglected altogether…it should not have a huge effect on the charge density you are concerned with…
- _get_conf_parameters(conf_file='hf.conf')[source]
- Parameters:
- conf_filestr
Path to
*.conffile to read from. If the file does not exist then the function will attempt to readhf.conffrom normal lookup storage locations, including (in order):ATLIBor~/atlib/~/or%USERPROFILE%/~/.phaseshifts/or%APPDATA%/phaseshifts‘./’
- Returns:
- Dictionary of Atorb.gen_input() keyword arguments.
- atlib = '~/atlib/'
- static calculate_Q_density(**kwargs)[source]
Execute the atomic structure calculation to obtain radial charge densities.
This method serves as the primary driver for the ‘atorb’ Fortran routine. It either generates a new input file based on an element symbol or validates an existing input file, then invokes the solver (hartfock) via libphsh.
The solver computes the radial wavefunctions
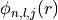 and
constructs the total spherical charge density .
and
constructs the total spherical charge density .- Parameters:
- **kwargsdict, optional
Keyword arguments passed to
gen_input()if element is provided.- elementstr or int
Target element. If provided, an input file is generated on-the-fly.
- inputstr
Path to an existing atorb input file. Ignored if element is provided.
- output_dirstr
Directory where the output file (containing phase shifts or charge densities) should be placed. Defaults to current directory.
- Returns:
- str
Path to the output file containing the calculated atomic charge densities.
- Raises:
- ValueError
If neither element nor input is specified.
- IOError
If the output directory cannot be created or accessed.
Examples
>>> Atorb.calculate_Q_density(input='atorb_C.txt') 18.008635 -33.678535 4.451786 -36.654271 1.569616 -37.283660 0.424129 -37.355634 0.116221 -37.359816 0.047172 -37.360317 0.021939 -37.360435 0.010555 -37.360464 0.005112 -37.360471 0.002486 -37.360473 0.001213 -37.360473 0.000593 -37.360473 0.000290 -37.360474 N L M J S OCC. 1 0 0 -1/2 1 2.0000 -11.493862 2 0 0 -1/2 1 2.0000 -0.788618 2 1 1 -1/2 1 0.6667 -0.133536 2 1 1 -3/2 1 1.3333 -0.133311 TOTAL ENERGY = -37.360474 -1016.638262
>>> Atorb.calculate_Q_density(element='H') 0.500007 -0.343752 0.152392 -0.354939 0.065889 -0.357254 0.028751 -0.357644 0.012732 -0.357703 0.005743 -0.357711 0.002641 -0.357712 0.001236 -0.357713 0.000587 -0.357713 0.000282 -0.357713 N L M J S OCC. 1 0 0 -1/2 1 1.0000 -0.229756 TOTAL ENERGY = -0.357713 -9.733932
- datalib = '~/.phaseshifts'
- property exchange
Returns the exchange correlation value, where 0.0=Hartree-Fock, 1.0=LDA or <float>=-alpha
- gen_conf_file(conf_file='hf.conf')[source]
- Parameters:
- conf_filestr
Filepath for conf output file (default: ‘hf.conf’).
- static gen_input(element, **kwargs)[source]
Generate the input file for the ‘atorb’ atomic structure solver.
This method constructs a formatted input file required by the Barbieri/Van Hove ‘atorb’ program (encapsulated in libphsh). It determines the electronic configuration of the specified element, handles noble gas core expansion, and sets up the radial grid and exchange-correlation parameters for the Dirac-Fock calculation.
- Parameters:
- elementstr or int
The chemical symbol (e.g., ‘Cu’) or atomic number (e.g., 29) of the target element.
- **kwargsdict, optional
Configuration options for the calculation:
- outputstr
Filename for the resulting charge density output (default: ‘at_<symbol>.i’).
- ngridint
Number of points in the logarithmic radial grid (default: 1000). Higher values provide better numerical resolution near the nucleus.
- relint or bool
Relativistic flag (default: 1). 1 (or True): Solve Dirac equations (includes spin-orbit coupling). 0 (or False): Solve non-relativistic Schrödinger equations.
- filenamestr
Filename for the generated input text file (default: ‘atorb_<symbol>.txt’).
- headerstr
Comment line at the top of the input file (default: auto-generated).
- methodstr
Exchange-correlation potential approximation (default: ‘0.d0’ for Hartree-Fock). ‘0.d0’: Hartree-Fock (HF). ‘1.d0’: Local Density Approximation (LDA). ‘-alpha’: X-alpha method (requires providing alpha value).
- relicfloat
Mixing parameter for the self-consistency cycle (default: 0.0).
- mixing_SCFfloat
Mixing parameter for Self-Consistent Field convergence (default: 0.5).
- tolerancefloat
Convergence tolerance for orbital eigenvalues (default: 0.0005 Hartrees).
- echint
Parameter for the exchange potential (default: 100).
- atorb_filestr or IO, optional
Override the destination for the generated input file. Accepts a file path or a file-like object (e.g., StringIO) for buffered writes.
- xnumfloat, optional
Extra numeric parameter used by EEASiSSS input formatting (default: 100).
- ifilint, optional
Flag to read vpert from vvalence. Only used when fmt=’rundgren’ or ‘eeasisss’ (default: 0).
- fmtstr, optional
Format of generated atorb input file; use ‘vht’ for Barbieri/Van Hove or ‘rundgren’/’eeasisss’ for EEASiSSS (default: ‘vht’).
- Returns:
- str
The path to the generated input file.
Notes
The resulting file allows libphsh to solve the radial Dirac equation:
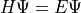
where density functional theory (DFT) or Hartree-Fock approximations define the potential.
- get_conf_parameters(conf_file='hf.conf')[source]
Public wrapper to read Atorb configuration parameters.
- static get_quantum_info(shell)[source]
Parse quantum numbers from a subshell string (e.g., ‘3d6’).
Decomposes a standard spectroscopic notation into the set of quantum numbers required for the radial equation solver. Handles the splitting of shells into spin-orbit coupled states (
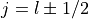 ).
).- Parameters:
- shellstr
Subshell string, e.g., ‘1s2’, ‘4f14’, ‘3d5’.
- Returns:
- tuple
(n, l, j_list, occ_list)
n (int): Principal quantum number.
l (int): Azimuthal quantum number.
j_list (list[float]): Total angular momentum values
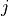 associated with this shell. For
associated with this shell. For 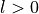 , this will be
[
, this will be
[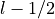 ,
, 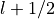 ].
].occ_list (list[float]): Occupancy for each
level.
Electrons are distributed according to statistical weight
(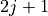 ).
).
Examples
>>> Atorb.get_quantum_info('3d6') (3, 2, [1.5, 2.5], [2.4, 3.6])
For a full ‘3d10’ shell:
>>> Atorb.get_quantum_info('3d10') (3, 2, [1.5, 2.5], [4.0, 6.0])
Here,
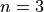 ,
, 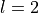 . The states are
. The states are 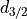 (occupancy 4)
and
(occupancy 4)
and 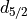 (occupancy 6).
(occupancy 6).
- property mixing_SCF
Returns the self-consisting field value
- property ngrid
Returns the number of points in the radial charge grid
- property rel
Returns boolean value of whether to consider relativistic effects
- property relic
Returns the relic value for calculation
- static replace_core_config(electron_config)[source]
Expand noble gas core abbreviations into full orbital strings.
The atorb solver requires explicit definitions for all orbitals starting from 1s. This function replaces ‘[Ar]’, ‘[Xe]’, etc., with their constituent subshells.
- Parameters:
- electron_configstr
Electronic configuration string, potentially containing noble gas cores (e.g., ‘[Ar] 4s2’).
- Returns:
- str
The fully expanded configuration string.
Examples
>>> Atorb.replace_core_config('[He] 2s1') '1s2 2s1'
- property tolerance
Returns the eigenvalue tolerance
- update_config(conf)[source]
- Parameters:
- confstr or dict
Either filepath to the user-specified
*.conffile containing the atomic charge density calculation parameters or else a dictionary of the keyword arguments to update.
- Raises:
- ValueError if
confis neither a str or dict instance.
- ValueError if
- userhome = '~/hf.conf'
- static validate_input_file(input_path)[source]
Validate a generated or user-supplied atorb input file.
Parses the input file using the phaseshifts.validation.atorb logic to ensure it meets the strict formatting and physical requirements of the solver.
- Parameters:
- input_pathstr
Path to the file to validate.
- Returns:
- AtorbInputModel
Parsed and validated representation of the input file.
- property xnum
Returns xnum value
- class phaseshifts.atorb.EEASiSSSAtorb(ifil=0, **kwargs)[source]
Bases:
Atorb- _get_conf_parameters(conf_file='~/atlib/hf.conf')[source]
- Parameters:
- conf_filestr
Path to
*.conffile to read from. If the file does not exist then the function will attempt to readhf.conffrom normal storage locations, including (in order):ATLIBor~/atlib/~/or%USERPROFILE%/~/.phaseshifts/or%APPDATA%/phaseshifts‘./’
- Returns:
- Dictionary of keyword arguments for
Atorb.gen_input().
- Dictionary of keyword arguments for
- static calculate_Q_density(elements=None, atorb_input='inputA', output_dir=None, **kwargs)[source]
- Parameters:
- elementslist of Element, int or str
Generate atorb input file on the fly. If the list is empty then the function will return an empty list.
- atorb_inputstr, optional
Specifies the path to the atorb input file. If
Nonethen a temporary file will be created.- output_dirstr, optional
Specifies the output directory for the at_*.i file generated. (default: :envar:`$ATLIB` or
~/atlib)- subroutinefunction, optional
Specifies the hartfock function to use (default: eeasisss_hartfock)
Warning
Do not modify the
subroutinevalue without good cause - here be dragons!
- Returns:
- List of filepaths to the calculated atomic charge density files.
Notes
This method implicitly calls
EEASiSSSAtorb.gen_input()for generating a suitable input file for the charge density calculations.For the patient, it may be worth having a initial one-time atomic charge density calculation for every element. This can be done like so:
from phaseshifts.elements import ELEMENTS, SERIES from phaseshifts.atorb import EEASiSSSAtorb # calculate chgden* files for EVERY element # and place them in the default location EEASiSSSAtorb.calculate_Q_density(elements=ELEMENTS)
Note
The above calculation is very useful and can be customised using a user generated
hf.conffile. For more details seeEEASiSSSAtorb.gen_conf_file()Examples
>>> from phaseshifts.elements import ELEMENTS, SERIES >>> from phaseshifts.atorb import EEASiSSSAtorb >>> >>> # generate hartfock input file for InGaAs >>> InGaAs = ['In', 31, 'Arsenic'] >>> input = '~/atlib/InGaAs.hf' >>> EEASiSSSAtorb.calculate_Q_density(elements=InGaAs, >>> atorb_input=input) >>> >>> # generate hartfock input file for all halogens >>> halogens = [e for e in ELEMENTS if SERIES[e.series] == 'Halogens'] >>> input = '$ATLIB/halogens.hf' >>> EEASiSSSAtorb.calculate_Q_density(halogens, atorb_file=input) >>> >>> # do likewise for all non-metals, but using hf.conf file parameters >>> series = 'Nonmetals' >>> non_metals = [e for e in ELEMENTS if SERIES[e.series] == series] >>> input = './nonmetals.hf' >>> EEASiSSSAtorb.calculate_Q_density(non_metals, atorb_file=input)
- gen_conf_file(conf_file='~/atlib/hf.conf')[source]
- Parameters:
- conf_filestr
Filepath for conf output file (default: ‘~/atlib/hf.conf’).
Examples
>>> from phaseshifts.atorb import EEASiSSSAtorb >>> atorb = EEASiSSSAtorb() # create an object instance >>> # create a config file in the default location >>> atorb.gen_conf_file()
- static gen_input(elements=None, atorb_file='inputA', **kwargs)[source]
- Parameters:
- elementslist
List of elements to include in the generated input file. Each element can be either the atomic number, symbol, name or an
phaseshifts.Elementinstance.- outputstr, optional
File string for atomic orbital output (default: ‘at_<symbol>.i’)
- ngridint, optional
Number of points in radial grid (default: 1000)
- relbool, optional
Specify whether to consider relativistic effects (default: True)
- atorb_filestr, optional
Name for generated input file (default: ‘inputA’)
- headerstr, optional
Comment at beginning of input file (default: None)
- methodstr or float, optional
Exchange correlation method using either 0.0=Hartree-Fock, 1.0=LDA, -alpha = float (default: 0.0)
- relicfloat, optional
Relic value for calculation (default: 0)
- mixing_SCFfloat, optional
Self consisting field value (default: 0.5)
- tolerancefloat, optional
Eigenvalue tolerance (default: 0.0005)
- xnumfloat, optional
??? (default: 100)
- ifilint, optional
flag to read
vpertarray fromvvalence- possibly redundant. Only used when fmt=’rundgren’ or ‘eeasisss’ (default: 0)
- Returns:
- Filename of input file once generated or else instance of StringIO
- object containing written input text.
Nonewill be returned if - the method failed to generate the input.
Notes
output can also be a StringIO() object to avoid saving to file.
Examples
>>> from phaseshifts.elements import ELEMENTS, SERIES >>> >>> # generate hartfock input file for InGaAs >>> InGaAs = ['In', 31, 'Arsenic'] >>> EEASiSSS.gen_input(elements=InGaAs, atorb_file='~/atlib/InGaAs.hf') >>> >>> # generate hartfock input file for all halogens >>> halogens = [e for e in ELEMENTS if SERIES[e.series] == 'Halogens'] >>> EEASiSSS.gen_input(halogens, atorb_file='$ATLIB/halogens.hf') >>> >>> # do likewise for all non-metals, but using hf.conf file parameters >>> non_metals = [e for e in ELEMENTS if SERIES[e.series] == 'Nonmetals'] >>> EEASiSSS.gen_input(non_metals, atorb_file='./nonmetals.hf')
- get_conf_parameters(conf_file='~/atlib/hf.conf')[source]
Public wrapper to read EEASiSSS-specific configuration parameters.
- property ifil
Returns flag for reading
vpertarray from filevvalence
- phaseshifts.atorb.eeasisss_hartfock(input_file)[source]
Lightweight wrapper around the EEASiSSS hartfock routine.
The underlying Fortran expects optional log/output arguments; this wrapper matches the single-argument call shape used by
Atorb.calculate_Q_density().
- phaseshifts.atorb.get_electron_config(element_obj)[source]
Extract the electronic configuration string from an element object.
Retrieves the standard ground-state configuration (e.g., ‘[Ar] 4s2 3d10 4p5’) from the backend-specific element object.
- Parameters:
- element_objobject
The element object returned by get_element.
- Returns:
- str
The electronic configuration string.
- phaseshifts.atorb.get_element(element: str, backend: Literal['mendeleev', 'elementy', 'periodictable'] | None = None) object[source]
Retrieve an element object from the specified chemical data backend.
This function abstracts the details of various chemistry libraries (mendeleev, elementy, periodictable) to provide a unified interface for accessing atomic properties like proton count (Z) and electronic configurations.
- Parameters:
- elementstr or int
The symbol (e.g., ‘Fe’) or atomic number (e.g., 26) of the element.
- backendstr, optional
The preferred backend library to use (‘mendeleev’, ‘elementy’, ‘periodictable’). If None, tries available backends in order.
- Returns:
- object
An object containing element data (attributes vary by backend but usually include protons, symbol, name).
- Raises:
- LookupError
If the element cannot be found in any available backend.
11.3.2. phaseshifts.conphas
conphas.py
Provides a native python version of the conphas (phsh3) FORTRAN program by W. Moritz, which is distributed as part of the SATLEED code (see “Barbieri/Van Hove phase shift calculation package” section) and can be found at: http://www.icts.hkbu.edu.hk/surfstructinfo/SurfStrucInfo_files/ leed/leedpack.html
The Conphas() class also provides a number of convenience functions (see docstrings below).
11.3.2.1. Examples
>>> from os.path import join
>>> from phaseshifts.conphas import Conphas
>>> con = Conphas(output_file=join('testing', 'leedph_py.d'), lmax=10)
>>> con.set_input_files([join('testing', 'ph1')])
>>> con.set_format('cleed')
>>> con.calculate()
- class phaseshifts.conphas.Conphas(input_files=None, output_file=None, formatting=None, lmax=10, v0_params=None, **kwargs)[source]
Bases:
objectClass Conphas
Notes
This work is based on the original conphas (phsh3) FORTRAN program by W. Moritz, which is distributed as part of the SATLEED code (see “Barbieri/Van Hove phase shift calculation package” section) and can be found at: http://www.icts.hkbu.edu.hk/surfstructinfo/SurfStrucInfo_files/ leed/leedpack.html
- __fix_path(file_path)
Fix escaped characters in filepath
- __set_data(data=None)
- _resolve_v0_params()[source]
Resolve Rundgren inner potential parameters for ViPErLEED output.
- Returns:
- tuple(float, float, float, float)
c0-c3 parameters; defaults to ViPErLEED’s documented values if none are provided. Raises a ValueError if an invalid v0_params configuration is supplied.
- _write_viper_output(file_handle, conpha, energy, n_phases)[source]
Write phase shifts in ViPErLEED PHASESHIFTS format.
- Parameters:
- file_handleIO
Open file handle for writing.
- conphalist
Continuous phase shifts per atom/site.
- energylist
Energies in eV as parsed from phasout.
- n_phasesint
Number of energy points to write.
- calculate()[source]
Calculates continuous phase shifts from input file(s).
Examples
>>> con = Conphas(output_file=r'testing/leedph_py.d', lmax=10) >>> con.set_input_files([r'testing/ph1']) >>> con.set_format('cleed') >>> con.calculate() L = 0 jump between 25.0 eV and 30.0 eV; IFAK = -1 L = 1 jump between 65.0 eV and 70.0 eV; IFAK = -1 L = 2 jump between 20.0 eV and 25.0 eV; IFAK = 1 jump between 80.0 eV and 85.0 eV; IFAK = 0 L = 3 L = 4 jump between 275.0 eV and 280.0 eV; IFAK = 1 L = 5 L = 6 L = 7 L = 8 L = 9 L = 10
- load_data(filename)[source]
Load (discontinuous) phase shift data from file
- Parameters:
- filestr
Path to phase shift file.
- Returns:
- tuple: (double, double, int, int, ndarray)
(initial_energy, energy_step, n_phases, lmf, data)
Notes
initial_energy is the starting energy of the phase shifts.
energy_step is the change in energy between consecutive values.
n_phases is the number of phase shifts contained in the file.
lmf is the maximum azimuthal quantum number considered.
data is a (2 x n_phases) array containing the phase shift data.
- read_datafile(filename)[source]
Read in discontinuous phase shift file
- Parameters:
- filenamestr
The path to the discontinuous phase shift file
- set_format(formatting=None)[source]
Set appropriate format from available options
- Parameters:
- formatstr, optional
The format identifier for different packages; can be ‘cleed’, ‘curve’, ‘viperleed’, or None.
11.3.3. phaseshifts.elements
Properties of the chemical elements.
Each chemical element is represented as an object instance. Physicochemical and descriptive properties of the elements are stored as instance attributes.
- Author:
- Version:
2013.03.18
11.3.3.1. Requirements
11.3.3.2. References
11.3.3.3. Examples
>>> from elements import ELEMENTS
>>> len(ELEMENTS)
109
>>> str(ELEMENTS[109])
'Meitnerium'
>>> ele = ELEMENTS['C']
>>> ele.number, ele.symbol, ele.name, ele.eleconfig
(6, 'C', 'Carbon', '[He] 2s2 2p2')
>>> ele.eleconfig_dict
{(1, 's'): 2, (2, 'p'): 2, (2, 's'): 2}
>>> sum(ele.mass for ele in ELEMENTS)
14659.1115599
>>> for ele in ELEMENTS:
... ele.validate()
... ele = eval(repr(ele))
11.3.4. phaseshifts.leed
Provides CLEED validator and Converter classes.
The CLEEDInputValidator() class provides a method for checking the input files for errors, whereas the Converter.import_CLEED() method allows importing CLEED input files as a MTZ_model class
- class phaseshifts.leed.CLEEDInputValidator[source]
Bases:
objectClass for validation of CLEED input files
- class phaseshifts.leed.CSearch(model_name, leed_command=None)[source]
Bases:
objectclass for csearch related data exchange
- class phaseshifts.leed.Converter[source]
Bases:
objectConvert different input into phaseshift compatible input
- static _load_structured_input(filename, yaml_loader=None)[source]
Load structured input (JSON or YAML).
- static _validate_structured_input(data)[source]
Validate cleedpy-style input if jsonschema is available.
- static cleedpy_to_inputs(filename, tmp_dir=None, yaml_loader=None, **kwargs)[source]
Convert cleedpy YAML to temporary bulk/slab .i files for phase-shift generation.
- Returns:
- tuple(str, str, dict)
Paths to generated bulk and slab input files, plus parsed metadata.
- static import_CLEED(filename, **kwargs)[source]
Imports CLEED input file and converts model to muffin-tin input.
- It assumes the following:
the basis vectors
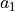 ,
, 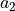 , &
, & 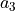 are
are
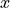 ,:math:y,:math:z cartezian coordinates
,:math:y,:math:z cartezian coordinatesif no
is found, the maximum  distance between
atoms multiplied by four is given.
distance between
atoms multiplied by four is given.the unitcell is converted from cartezian to fractional coordinates
atom sites are converted from Angstrom to Bohr units
additional info from the phase shift filename is provided by splitting the ‘_’ chars:
First string segment is element or symbol, e.g. Ni
Second string segment is the oxidation (valence), e.g. +2
lines with
rm:provide the radii dictionary of the individual phase shifts, whereas lines starting withlmax:provide the lmax dictionary for individual phase shifts. Additionally, the valency can be given in lines starting withox:. Both bulk and surface input files will be searched for these.if no
rm:found for that species, the atomic radius is used for zero valence, otherwise the covalent radius is used.if no lmax values are found for the specific phase shift, then the global value will be used instead.
if no valence values are found for the specific phase shift, then the guessed oxidation value from the phase shift filename is used instead. However, if the oxidation state cannot be parsed using the filename then a default oxidation state of zero is used.
Additional information can, however, be provided using ‘phs:’ at the start of a line within the input file and may have the following formats:
“phs: c <float> nh <int> nform <int> exchange <float>”
“phs: <phase_shift> valence <float> radius <float>”
The identifiers
exchange,nform,valenceandradiusmay be abbreviated toexc,nf,valandrad, respectively.Information given in this manner overrides any other input.
- Parameters:
- filenamestr
Path to input file.
- Returns:
- phaseshifts.model.MTZ_model
- Raises:
- IOErrorfilename invalid
- ValueErrorbad formatting of input
- static import_cleedpy_input(filename, superstructure=True, yaml_loader=None, **kwargs)[source]
Parse a cleedpy-style structured input file (JSON or YAML) into bulk and slab MTZ models.
- Parameters:
- filenamestr
Path to cleedpy YAML input.
- superstructurebool, optional
If True, apply the superstructure_matrix (m1/m2) to the in-plane basis vectors to construct the surface cell. Defaults to True.
- Returns:
- tuple(model.MTZ_model, model.MTZ_model, dict)
(bulk_model, slab_model, metadata)
Notes
This provides a bridge from cleedpy’s YAML geometry to the Barbieri/Van Hove phase-shift workflow by constructing equivalent muffin-tin models.
11.3.5. phaseshifts.model
model.py
Provides convenience functions for generating input and calculating atomic charge densities for use with the Barbieri/Van Hove phase shift calculation package.
- class phaseshifts.model.Atom(element, coordinates=[0.0, 0.0, 0.0], **kwargs)[source]
Bases:
objectAtom class for input into cluster model for muffin-tin potential calculations.
- exception phaseshifts.model.CoordinatesError(msg)[source]
Bases:
ExceptionCoordinate exception to raise and log duplicate coordinates.
- class phaseshifts.model.MTZ_model(unitcell, atoms, **kwargs)[source]
Bases:
ModelMuffin-tin potential Model subclass for producing input file for muffin-tin calculations in the Barbieri/Van Hove phase shift calculation package.
- _load_input_file(filename)[source]
- Parameters:
- filenamestr
The path of the input file (e.g. cluster*.i or slab.i)
- Raises:
- IOErrorexception
If the file cannot be read.
- TypeErrorexception
If a input line cannot be parsed correctly.
- calculate_MTZ(mtz_string='', **kwargs)[source]
- Parameters:
- atomic_filestr
The path to the atomic input file. If this is omitted the default is generate one using the MTZ_model.gen_atomic() method.
- cluster_filestr
The path to the cluster input file which may be a bulk or slab model.
- slabint or bool
Determines whether the MTZ calculation is for a slab model (True). The default is a bulk calculation.
- outputdict
Dictionary output of ‘mtz’ - muffin-tin potential & ‘output_file’ - the path to the MTZ output file.
- Returns:
- output_fileslist(str)
Paths to the MTZ output file.
- create_atorbs(**kwargs)[source]
- Returns:
- output_filesdict
Dictionary list of atorb*.i input files for the Atorb class to calculate the charge density from.
- gen_atomic(**kwargs)[source]
- Parameters:
- input_dirstr
Input directory where at*.i files are kept.
- input_filestuple
List of input files to generate atomic input file from.
- output_filestr
The filename of the resulting atomic*.i output file, which is simply a superimposed set of the radial charge densities from the individual input files.
- Returns:
- output_filestr
Returns the output file path string.
- Raises:
- IOErrorexception
If either input directory or files do not exist.
Notes
If ‘input_files’ is not given then the default list of input files are inferred from the list of atoms in the model.
- gen_input(**kwargs)[source]
- Returns:
- filename on success
- Raises:
- CoordinatesErrorexception
if the model atoms have duplicate coordinates and the ‘pos_check’ kwarg is set to True.
- load_from_file(filename)[source]
- Parameters:
- filenamestr
The path of the input file (e.g. cluster*.i or slab.i)
- Raises:
- IOErrorexception
If the file cannot be read.
- TypeErrorexception
If a input line cannot be parsed correctly.
- set_nform(nform)[source]
Sets form of muffin-tin calculation
- Parameters:
- nformint or str
This governs the type of calculation, where nform can be:
1. “cav” or 0 - use Cavendish method
2. “wil” or 1 - use William’s method
3. “rel” or 2 - Relativistic calculations
- set_slab_c(c)[source]
Set the maximum height of the slab in Angstroms.
If this is much larger than the bulk c distance then there will be a large vacuum and therefore should be used when calculating a thin slab rather than a bulk muffin-tin potential.
Examples
For Re the bulk c distance is 2.76Å, whereas a possible slab c distance could be ~10Å.
- class phaseshifts.model.Model(unitcell, atoms, **kwargs)[source]
Bases:
objectGeneric model class.
- _nineq_atoms()[source]
- Returns:
- nineq_atoms, element_dicttuple
- nineq_atomsThe estimated number of inequivalent atoms based on
the valence and radius of each atom.
- element_dicta dictionary of each element in the atom list where
each element contains an atom dictionary of ‘nineq_atoms’, ‘n_atoms’ and a complete ‘atom_list’
- add_atom(element, position, **kwargs)[source]
Append an Atom instance to the model
- Parameters:
- elementstr or int
Either an element name, symbol or atomic number.
- positionlist(float) or ndarray
(1x3) array of the fractional coordinates of the atom within the unit cell in terms of the lattice vector a.
- property atoms
Returns a list of atoms within this model
- check_coordinates()[source]
Check for duplicate coordinates of different atoms in model.
- Raises:
- CoordinateErrorexception
If duplicate positions found.
- property elements
Returns a list of unique elements within the model
- property name
Returns the model name
- class phaseshifts.model.Unitcell(a, c, matrix_3x3, **kwargs)[source]
Bases:
objectUnitcell class
- set_a(a)[source]
- Parameters:
- a: float
The magnitude of the in-plane lattice vector in Angstroms
Notes
To retrieve a in terms of Angstroms use ‘unitcell.a’, whereas the internal parameter ‘unitcell._a’ converts a into Bohr radii (1 Bohr = 0.529Å), which is used for the muffin-tin potential calculations in libphsh (CAVPOT subroutine).
- set_c(c)[source]
- Parameters:
- cfloat
The magnitude of the in-plane lattice vector in Angstroms
Notes
To retrieve c in terms of Angstroms use ‘unitcell.c’, whereas the internal parameter ‘unitcell._c’ converts c into Bohr radii (1 Bohr = 0.529Å), which is used for the muffin-tin potential calculations in libphsh (CAVPOT subroutine).
11.3.6. phaseshifts.phsh
phsh.py - quickly generate phase shifts
Orchestrates the full phase shift calculation workflow for LEED (Low-Energy Electron Diffraction) and related surface science applications, following the Barbieri/Van Hove methodology.
This module provides a command-line interface and Python API for generating phase shift files suitable for input into LEED-IV programs such as SATLEED and CLEED. It automates the process of: - Atomic charge density calculation (Dirac-Fock/Hartree-Fock) - Muffin-tin potential generation - Phase shift calculation (relativistic/non-relativistic) - Removal of phase discontinuities (pi-jumps) - Output formatting for LEED analysis
11.3.6.1. Scientific Context
Phase shifts are central to electron scattering theory and LEED surface structure analysis. The workflow implemented here follows the Barbieri/Van Hove package, ensuring that all physical and computational steps are performed in the correct sequence, with appropriate file formats and conventions.
11.3.6.2. References
- `Barbieri, G., & Van Hove, M. A. (1979). “Phase shift calculation package for LEED.” Surf. Sci. 90, 1-25.
Moritz, W. (SATLEED code): http://www.icts.hkbu.edu.hk/surfstructinfo/SurfStrucInfo_files/leed/leedpack.html
See also: https://phaseshifts.readthedocs.io/en/latest/phshift2007.html
11.3.6.3. Examples
phsh.py -i *.inp -b *.bul -f CLEED -S phase_dir
- exception phaseshifts.phsh.CLIError(msg)[source]
Bases:
ExceptionGeneric exception to raise and log different fatal errors.
- class phaseshifts.phsh.Wrapper(**kwargs)[source]
Bases:
objectClass for automating the Barbieri/Van Hove phase shift calculation workflow.
This class provides methods to generate phase shift files from atomic and cluster input data, handling all intermediate steps: atomic charge density, muffin-tin potential, phase shift calculation, phase-jump removal, and output formatting. It supports both Python and Fortran backends, and is compatible with SATLEED and CLEED file formats.
11. Scientific Rationale
Automating the workflow ensures reproducibility and correct sequencing of physical and computational steps, as required for LEED surface structure analysis.
References
`Barbieri, G., & Van Hove, M. A. (1979). “Phase shift calculation package for LEED.” Surf. Sci. 90, 1-25.
Moritz, W. (SATLEED code): http://www.icts.hkbu.edu.hk/surfstructinfo/SurfStrucInfo_files/leed/leedpack.html
See also: https://phaseshifts.readthedocs.io/en/latest/phshift2007.html
- static _calculate_bulk_mtz(bulk_mtz, bulk_file, bulk_atomic_file, tmp_dir, bulk_model_name, verbose)[source]
- static _calculate_slab_mtz(slab_mtz, slab_file, slab_atomic_file, tmp_dir, slab_model_name, bulk_mtz, verbose)[source]
- static _copy_files(files, dst, verbose=False)[source]
Copy list of files into destination directory.
- static _generate_phase_shifts(slab_mtz, mufftin_filepath, phasout_filepath, dataph_filepath, filepath, out_format, phasout_files, phaseshifts, lmax_dict, kwargs)[source]
- static autogen_from_input(bulk_file, slab_file, tmp_dir=None, model_name=None, **kwargs)[source]
Generate phase shifts from slab/cluster and bulk input files, following the Barbieri/Van Hove workflow.
This method automates the full sequence described in: - `Barbieri, G., & Van Hove, M. A. (1979). “Phase shift calculation package for LEED.”
Surf. Sci. 90, 1-25. <https://doi.org/10.1016/0039-6028(79)90470-7>`_
Moritz, W. (SATLEED code): http://www.icts.hkbu.edu.hk/surfstructinfo/SurfStrucInfo_files/leed/leedpack.html
See also: https://phaseshifts.readthedocs.io/en/latest/phshift2007.html
Steps: 1. Atomic charge density calculation (Dirac-Fock/Hartree-Fock) 2. Muffin-tin potential generation 3. Phase shift calculation (relativistic/non-relativistic) 4. Removal of phase discontinuities (pi-jumps) 5. Output formatting for LEED analysis
- Parameters:
- bulk_filestr
Path to the bulk MTZ or CLEED input file.
- slab_filestr
Path to the slab/cluster MTZ or CLEED input file.
- tmp_dirstr, optional
Temporary directory for intermediate files.
- model_namestr, optional
Name of the model.
- storebool or str, optional
Whether to keep generated files and where to store them.
- formatstr, optional
Output format for phase shift files (‘cleed’, ‘curve’, etc.).
- rangetuple(float, float, float), optional
Energy range for phase shift calculation (start, stop, step in eV).
- Returns:
- output_fileslist of str
List of generated phase shift output filenames.
- phaseshifts.phsh._generate_atomic_orbitals(bulk_file, slab_file, tmp_dir=None, verbose=False)[source]
Generate atomic charge density files for elements in bulk and slab inputs.
- phaseshifts.phsh.main(argv=None)[source]
Run the command-line interface for phase shift generation.
Parses user arguments, sets up the workflow, and executes the full Barbieri/Van Hove phase shift calculation sequence. Supports options for input files, output formatting, energy range, and intermediate file management.
- Parameters:
- argvlist of str, optional
Command-line arguments (default: sys.argv).
- Returns:
- int
Exit code (0 for success).
References
`Barbieri, G., & Van Hove, M. A. (1979). “Phase shift calculation package for LEED.” Surf. Sci. 90, 1-25.
Moritz, W. (SATLEED code): http://www.icts.hkbu.edu.hk/surfstructinfo/SurfStrucInfo_files/leed/leedpack.html
See also: https://phaseshifts.readthedocs.io/en/latest/phshift2007.html