15. APPENDIX II: Elastic Electron-Atom Scattering in Solids and Surface Slabs - User Guide
John Rundgren
KTH Royal Institute of Technology,
Theoretical Physics AlbaNova,
SE-10691 Stockholm,
Sweden
15.1. Acknowledgement notice
Please acknowledge use of the EEASiSSS package by citing the following:
J. Rundgren, Phys. Rev. B, 68, 125405 (2003).
J. Rundgren, Phys. Rev. B, 76, 195441 (2007).
15.2. Contact
John Rundgren: jru@kth.se
15.3. Overview
The EEASiSSS program is characterized by three features:
Muffin-tin radii
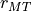 and charge content
and charge content 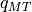 per
MT sphere are calculated inside the program without any exterior
start values.
per
MT sphere are calculated inside the program without any exterior
start values.The inner potential is spatially flat. It gets an energy variation due to the use of Kohn-Sham and Hedin-Lundqvist local density approximation to the exchange-correlation interaction of incident-electron and crystal electron gas [1] [2] . In consequence
and show
weak energy variations.Coulomb potential and electron density are generated by superposition of free-atom potentials (default) or extracted from WIEN2k’s FLAPW program packet by means of an ASA routine [3] . FLAPW is full-potential linearized augmented plane wave; ASA is atomic sphere approximation.
WIEN2k is a licensed program packet and the ASA routine is WIEN2k property. Public use of ASA requires that the routine be acknowledged by the WIEN2k team.
The variation of the inner potential versus incident energy puts a particular programming condition on the LEED program applying EEASiSSS. Calculated phase shifts, describing electron scattering inside the crystal, are referred to the in-crystal energy:
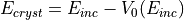
Where 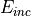 is the incident energy.
is the incident energy.
EEASiSSS provides an interstitial energy file 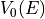 to be read-in by
the LEED program. A selection math:V_0 = const is provided for LEED programs
not equipped with
to be read-in by
the LEED program. A selection math:V_0 = const is provided for LEED programs
not equipped with 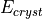 code.
code.
K. Heinz et al. have shown by LEED experiment that the use of
resolves structural parameters with precision 0.01 Angstroms [4] . Selection
was the winner in an accuracy competition between various
phase shift methods arranged by V.B. Nascimento et al. in a LEED investigation
on the (001) surface of calcium strontium ruthenate [5] . Recently
has been used in several complex LEED cases, for instance,
magnetite [6] and polyaromatic trithiolate adsorbed on Cu(111) [7] .
The input data to an EEASiSSS calculation are illustrated by an input template on the GaAs(110)-(1x1) surface slab structure [8] .
15.4. EEASiSSS calculation
The EEASiSSS package performs the calculation of the phase shifts in two steps:
The atomic charge densities of each element in the model is calculated using the Hartree-Fock method by the hf.py program using a variant of Eric Shirley’s
hartfock()subroutine.The phase shifts are then calculated from the generated charge density files and an EEASiSSS input file describing the model geometry and calculation parameters.
Although these steps are taken care of automatically through the phsh script,
two utility programs - hf.py and eeasisss.py, respectively - are installed as
part of the phaseshifts package.
15.4.1. hf.py
A Python command line script is installed as part of the phaseshifts package which can be used to calculate atomic charge densities for different elements.
15.4.1.1. Syntax
The command line calling convention for the hf.py program is:
hf.py -i <input> [-a <chgden_dir> -o <log_file>]
The options are described below:
-i <input>Specifies the path to the atomic orbital input file. Default is ‘./InputA’.
-a <chgden_dir>Optional argument which specifies the output directory to place the calculated atomic charge density
chgden*files into. If no argument is given, theATLIBenvironment variable is used as the output directory, else ‘~/atlib’ path is used. Use ‘.’ for<chgden_dir>to place the output files into the current working directory.-o <log_file>Optionally redirects the calculation log to the file specified by
<log_file>. The default is to print tostdoutif the argument is omitted.
Note
The hf.py script should be installed on the system PATH
and therefore executing the program using the absolute file path should
not be needed.
An example input file can be found under examples/EEASiSSS/GaAs/inputA and covers the format of the atomic orbital input file.
The charge density output files for each element in the form: chgden<symbol>
15.4.2. eeasisss.py
15.4.2.1. Syntax
The eeasisss.py Python script can be called using the following syntax:
eeasisss.py -i <input> [-o <output_dir> -a <chgden_dir> -l <log_file>]
The command line options for calling the program are:
-i <input>Specifies the path to the EEASiSSS model input file. A complete description of the format is given below. Default is ‘./inputX’.
-l <log_file>Name of log file. Default is
stdoutif-largument is not specified.-o <output_dir>path to log file, phase shifts, and selected intermediate files. Default is ‘./result/’.
-a <chgden_dir>path to charge density files and to intermediate electrostatic MT charge file(s). Default, in order, is
ATLIBenvironment variable, then the path given by ‘~/atlib/’.
15.5. EEASiSSS input
What follows is an example of EEASiSSS applied to GaAs(110)-(1x1) surface structure.
1STRUCTURE:
2GaAs110
3$WIEN2kpath/GaAs110
41.889726 |FactorDefiningBohrInside [1.889726, WhenUnitCell&PositionsAreIn(A)].
5 3.997980 0.000000 0.000000
6 0.000000 5.653998 0.000000
7 0.000000 0.000000 15.610722
812 |#IneqAt
91 33 1 1 0.0,0.0,0.0 'y' |#EqAt,id(1:3),rMTmin,rMTmax,cls(eV),BmSel
10 0.000000 0.000000 0.000000 |x y z AtPos
111 31 2 8 0.0,0.0,0.0 'y'
12 1.998990 4.360516 0.686750
131 31 3 12 0.0,0.0,0.0 'y'
14 0.000000 1.193521 2.159887
151 33 4 2 0.0,0.0,0.0 'y'
16 1.998990 2.607020 2.189891
171 31 5 10 0.0,0.0,0.0 'y'
18 1.998990 4.020520 4.173879
191 33 6 3 0.0,0.0,0.0 'y'
20 0.000000 5.434019 4.173879
211 31 7 7 0.0,0.0,0.0 'y'
22 0.000000 1.193521 6.172869
231 33 8 4 0.0,0.0,0.0 'y'
24 1.998990 2.607020 6.172869
251 31 9 9 0.0,0.0,0.0 'y'
26 1.998990 4.020520 8.171859
271 33 10 5 0.0,0.0,0.0 'y'
28 0.000000 5.434019 8.171859
291 31 11 11 0.0,0.0,0.0 'y'
30 0.000000 1.193521 10.170849
311 33 12 6 0.0,0.0,0.0 'y'
32 1.998990 2.607020 10.170849
33OPTIONS:
34'S' |VCoul :'S'=FreeAtomSuperpos/'W'=WIEN2k.
35'E' |Vxc :'E'=EnergyDependent V0(E)/'C'=constant V0.
36'n' |VMadl : MadelungElectrostaticPotential? 'yes'/'no'.
37'D' |MTrad :'D'=DEopt/'M'=MidptNNdist/'I'=IntersecNNpot.
38'n' |SpinPS: print_SpinUp&DownPhaseShifts? 'yes'/no'.
39'nn' |print_RhoPot? 'yes'/'no' and 'yes'/'no' (ie=1 default).
40'n' |print_WaveFunction? 'yes'/'no' (Einc fixed).
41'n' |print_TotalCrossSection sigma? 'yes'/'no' (Einc varying).
42'n' |print_log10(DsigmaDomega)? 'yes'/'no' (Einc fixed).
43'n' |print_DsigmaDtheta? 'yes'/'no' (Einc fixed).
44'n' |print_Sherman? 'yes'/'no' (Einc fixed).
452.,4.,2. |energy grid: E1,E2,DE (start, end, mesh at start).
4606 |lmax
470.625d0 |rMTweight in interval (0.,1.), weight of rerr vs verr (see README).
480.075 |maximum verr (eV), accuracy of Vtot continuity at rMT(:).
49DIFFERENTIAL EVOLUTION METHOD:
501.d-06 | VTR (pot. step in eV).
519 0.8d0 0.5d0 | NPfac,F_XC,CR_XC
520 1 0 | method
532 0.8d0 | strategy,F_CR
5410000 0 | itermax,refresh
15.5.1. Comments on example input file
15.5.1.1. STRUCTURE Section
Line 1: Title of the surface structure; output files are named by the title following the designation convention of WIEN2k.
Line 2: Active with WIEN2k, path to charge-density and potential data.
Line 3: Factor defining length in Bohr units inside the program; it is 1.889726 when unit cell and atomic positions are given in Angstroms. Energy units inside are Rydbergs.
Line 4-6: Unit cell basis vectors a 1, a 2 and a 3, respectively, in Angstroms.
Line 7:
nieq= number of inequivalent atoms.Line 8: contains eight items, which are ordered as follows:
neq= number of equivalent atoms (= 1 in the template);Z= atomic number (i.e. 31 for Ga, 33 for As);identification number of atom, consecutive i=1,nieq.
When selection
Vcoul=='S': 4th item not used, can be set to zero; When selectionVcoul=='W': identification number in WIEN2k calculation, i.e. the integer of a WIEN2k permutation with value i=1,nieq.0in the template corresponds to 0.5 Bohr inside.0in the template corresponds to a value set by the charge density input.core level shift of atomic potential in eV.
print this particular phase shift?
'yes'/'no'.Note
Items 5, 6 and 7 are beta quantities that were provided for computer experiments with EEASiSSS.
Line 9: Atomic positions x,y,z of which x,y are parallel to surface plane and z increases with depth below surface. When
neqon line 8 is > 1, line 9 is replaced byneqlines of atomic positions.15.5.1.2. OPTIONS Section
VCoulwhich can be either:'S'for Free Atom Superposition'W'for WIEN2k.With
'S'atomic electron density and Coulomb potential are generated by superposition of free-atom charge densities. Selection'W', extracts the charge densities from a WIEN2k calculation.Vxcwhich can be either:'E'for Energy Dependent'C'for constantWith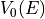 is
calculated from Kohn-Sham theory via the Hedin-Lundqvist local density
approximation; see figure README.V0vsE.ps.
With
is
calculated from Kohn-Sham theory via the Hedin-Lundqvist local density
approximation; see figure README.V0vsE.ps.
With 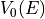 ,
,
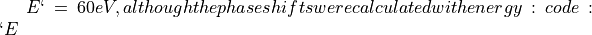 variable.
variable.
'E'the exchange-correlation potential'C'the inner potential is put equal toLine 3:
VMadl- a flag for whether the Madelung potential is used and can be either code:’yes’/’no’. Selection code:’n’ corresponds to current LEED. Selection'y'generates the Madelung (electrostatic) potential at a particular atom from the infinite collection of neighboring atoms.The literature on electrostatic potentials is incomplete for LEED investigations; required is a theory for semi-crystals z > 0 and for various distributions of top-layer charges.
In EEASiSSS an improvised surface description is used, cf. comments in subroutine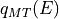 .
.
ProcRoySoc_ElectrostaticPotential. SelectionVMadl='y'requires that selectionVMadl='n'has been executed beforehand to produce charge content per MT sphereMTradhas three options:'D' = DEopt'M' = MidptNNdist'I' = IntersecNNpot.Selection
'D'is recommended: Differential Evolution method determines MT spheres from three conditions: that non-overlapping MT spheres are as large as possible; that MT potentials are continuous to a flat inner potential; and that all MT potentials are energy-shifted by the same amount.Old-fashioned selections
'M'and'I'are at user’s service:'M'sizes MT spheres approximately to halve next-neighbour distance, and'I'searches points on atom-to-atom lines where the next-neighbour potentials intersect.Line 5: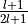 and
and 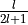 , respectively
(
, respectively
(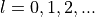 ).
).
SpinPSoption to print SpinPhaseShift?'yes'/no'. With'y'files are printed using designations .su. and .sd. for spin up and down, respectively. Relativistic spin-less phase shifts labelled .sl. are calculated from .su. and .sd. as linear averages with weightsLine 12: Energy grid. The present program version uses a uniform energy grid of mesh 2 eV. By uncommenting lines in subroutine EnergyGrid one can create non-uniform grids getting gradually sparser at energies above a convenient limit, say, 80 eV.
Line 14: Selection of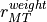 .
.
Two errors are calculated,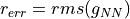 and
and
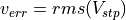 , from two concurrent quantities
, from two concurrent quantities
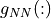 = gap between next-neighbor MT spheres (in Bohr),
= gap between next-neighbor MT spheres (in Bohr),
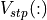 = discontinuity of potential at
= discontinuity of potential at 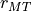 (in eV),
where (:) symbolizes set of atoms,
(in eV),
where (:) symbolizes set of atoms, 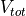 is total potential of
MT sphere, and
is total potential of
MT sphere, and 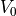 is interstitial potential. Using rMTweight the errors
are weighted together to a single fitness value guiding the optimization
by the Differential Evolution method.
is interstitial potential. Using rMTweight the errors
are weighted together to a single fitness value guiding the optimization
by the Differential Evolution method.
where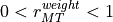
It is noted that fitness combines quantities of different units of measurement. The mix of units seems meaningful when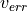 and
and
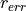 harmonize with the resolution of a LEED investigation in terms
of voltage and interatomic distance.
harmonize with the resolution of a LEED investigation in terms
of voltage and interatomic distance.
Line 15: accuracy (in eV) of zeroth order potential continuity at MT sphere radius. Physically available and acceptable values of accuracy and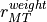 are studied in the log file.
are studied in the log file.
15.5.1.3. DIFFERENTIAL EVOLUTION METHOD Section
Selection of parameters from reference: R. Storn and K. V. Price, J. Global Optimization 11, 341-59 (1997); http://www1.icsi.berkeley.edu/~storn/code.html.
NPfacanditermaxhave high or low values when the number of atoms is high or low; fitness and number of iterations are inspected in the EEASiSSS log.